CELEBRATING 32 YEARS OF SELFLESS SERVICE
Total Consultations
Surgeries and Procedures
Totally free of charge to all
Specialities

Cardiology
The Department of Cardiology began functionaing in the Sri Sathya Sau Institute of Higher Medical Sciences …

CTVS
The Department of Cardio-Thoracic and Vascular Surgery (CTVS) has been rendering effective ..

Ophthalmology
The Department of Ophthalmology has been providing advanced eye care since 1994. The department has …

Plastic Surgery
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Gastroenterology
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Urology
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Orthopaedics
The department was opened at Sri Sathya Sai Institute of Higher Medical Sciences (SSSIHMS), Prasanthigram, as …
SERVICES

Radiology
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Anaesthesiology
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Nursing
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Laboratory Services
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Dietary Services
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …

Blood Bank
The department began functioning on the day the hospital was inaugurated, 22-Nov 1991, wit …
WHAT IS NEW
STATE-OF-THE-ART CATHETERISATION LABORATORY INAUGURATED

With the Divine Blessings of Swami, a state-of-the-art Catheterisation Laboratory worth Rs. 4.2 Crore was inaugurated by Sri RJ Rathnakar (Managing Trustee, SSSCT) along with Trustees Sri K Chakravarthi, Sri SS Naganand and Sri Nimish Pandya (AIP, SSSSO) at the Cardiology Department of the Sri Sathya Sai Institute of Higher Medical Sciences, Prasanthigram today. Dr Gurumurthy (Director, SSSIHMS-PG), Dr V.R Iyer (HoD, Cardiology) and other senior staff were present on the occasion.
Cath Lab is a specialised facility used to provide minimal invasive advanced cardiac procedures to diagnose and treat cardiovascular diseases. Philips Azurion Cath Lab is the Next-Gen image-guided intuitive therapy platform, that has unique features like Dynamic Coronoray Roadmap, Digital Subtraction Angiography, reduced radiation dose and total control of the Cathlab at table side to name a few. These features make the system more efficient and hence patient diagnosis and treatment easier.
The Advanced Cath Lab is the second major addition to SSSIHMS-PG this year. Just a month ago a new Heart Lung Machine was inaugurated at the hospital. Sri RJ Rathnakar said that in Swami’s hospital we have the best doctors and the best equipment to provide the best of the patient care totally free of charge to all. Swami’s hospitals stand as beacons of hope to humanity especially for the poor and the needy. As the trustees of the Sri Sathya Sai Central Trust it is our commitment to see the institutions established by Bhagawan Baba grow and prosper with every passing day and provide better care to the people.
ADVANCED HEART LUNG MACHINE INAUGURATED AT SSSIHMS, PRASANTHIGRAM

With the Blessings of Bhagawan Sri Sathya Sai Baba an advanced Heart Lung Machine worth Rs. 1.18 crore was inaugurated at Cardio Thoracic Vascular Surgery department of the Sri Sathya Sai Institute of Higher Medical Sciences, Prasanthigram, Puttaparthi.
The Terumo Advanced Perfusion System 1 Heart Lung Machine (HLM) was inaugurated by the Managing Trustee of Sri Sathya Sai Central Trust, Sri R. J. Rathnakar in the presence of the Director of SSSIHMS, Prasanthigam, Dr. Gurumurthy, the Head of Department of Cardio Thoracic Vascular Surgery Dr. Neelam Desai and other senior staff members of the hospital. A Heart Lung Machine (HLM) is indispensable equipment used in Cardiac Surgeries. The HLM takes over the function of a human heart and maintains blood circulation when open heart/bypass surgeries are performed. It also consists of an oxygenator, which takes over the function of the lungs. Terumo Heart Lung Machines have been used at SSSIHMS since inception. The current acquisition is the latest version of Terumo Heart Lung Machines, the Terumo® Advanced Perfusion System 1. This is an interactive system, whose quality and technology features assist in providing the best possible patient outcomes.
A NEW STATE-OF-THE-ART O.C.T MACHINE INAUGURATED AT SSSIHMS, PRASANTHIGRAM

With the Blessings of Bhagawan Sri Sathya Sai Baba, a new state-of-the-art Optical Coherence Tomography (OCT) machine worth Rs. 60 lakh was inaugurated at the Sri Sathya Sai Institute of Higher Medical Sciences, Prasanthigram, Puttaparthi.
The OCT machine was inaugurated by the Managing Trustee of SSSCT, Sri R J Rathnakar in the presence of the Director of SSSIHMS, Prasanthigam, Dr. Gurumurthy, the Head of Department of Ophthalmology, Dr. Anuj Sharma and other senior staff members of the hospital. Optical coherence tomography (OCT) is a non-invasive imaging method that uses reflected light to create pictures of the back of your eye.
It can be used to diagnose and manage diseases like diabetes-related retinopathy, glaucoma etc. Manufactured by Carl Zeiss the OCT machine has many advanced features like Enhanced Depth Imaging (EDI) through which the machine can analyse beyond the layers of the retina.Also the system comes with a Fast Track feature, which helps in reducing the eye motion artifacts and thereby improving the accuracy of the diagnosis.
TWO DNB TRAINEES OF SSSIHMS, PRASANTHIGRAM RECEIVE GOLD MEDALS

Two DNB Trainees of the Sri Sathya Sai Institute of Higher Medical Sciences, Prasanthigram, Puttaparthi have secured the National Board of Examinations, Gold Medals for the year 2020. Dr. Yogi Sundar Rao Chaganti topped in the Cardio Vascular Thoracic Surgery stream, while Dr. Anuj Dumra came first in the Urology stream. The Urology Gold medal is named after Dr. H S Bhatt, considered a doyen of Urology in India, who had set up the Department of Urology at the SSSIHMS, Prasanthigram.
This is the first time that the hospital has received two gold medals in the same year.
Speaking on the occasion the Director of the SSSIHMS, Prasanthigram, Dr Gurumurthy said that the two gold medals in the same year were a testament to the high quality training provided by the hospital. “The SSSIHMS, Prasanthigram has the state-of-the-art equipment along with skilled manpower, which makes it the best place for any student trying to leave a mark in the their respective specialties,” Dr. Gurumurthy said. “As the patient care has imbibed principles of health care as affirmed by Bhagawan Sri Sathya Sai Baba, the trainees apart from the technical skills also learn the soft skills of treating the patients with love and compassion. This makes Bhagawan Baba’s hospital unique and different from other institutions. Here credit goes to the Sri Sathya Sai Central Trust, which has continued to provide support to the hospital in every way,” Dr Gurumurthy said.
Both Dr. Anuj Dumra and Dr Yogi Chaganti conveyed their gratitude first to their parents to have helped them to reach the place they are today. ” In any walk of life the blessings of the parents is of great importance and it is with their efforts and sacrifices that we both are where we are,” they said.
ACADEMICS
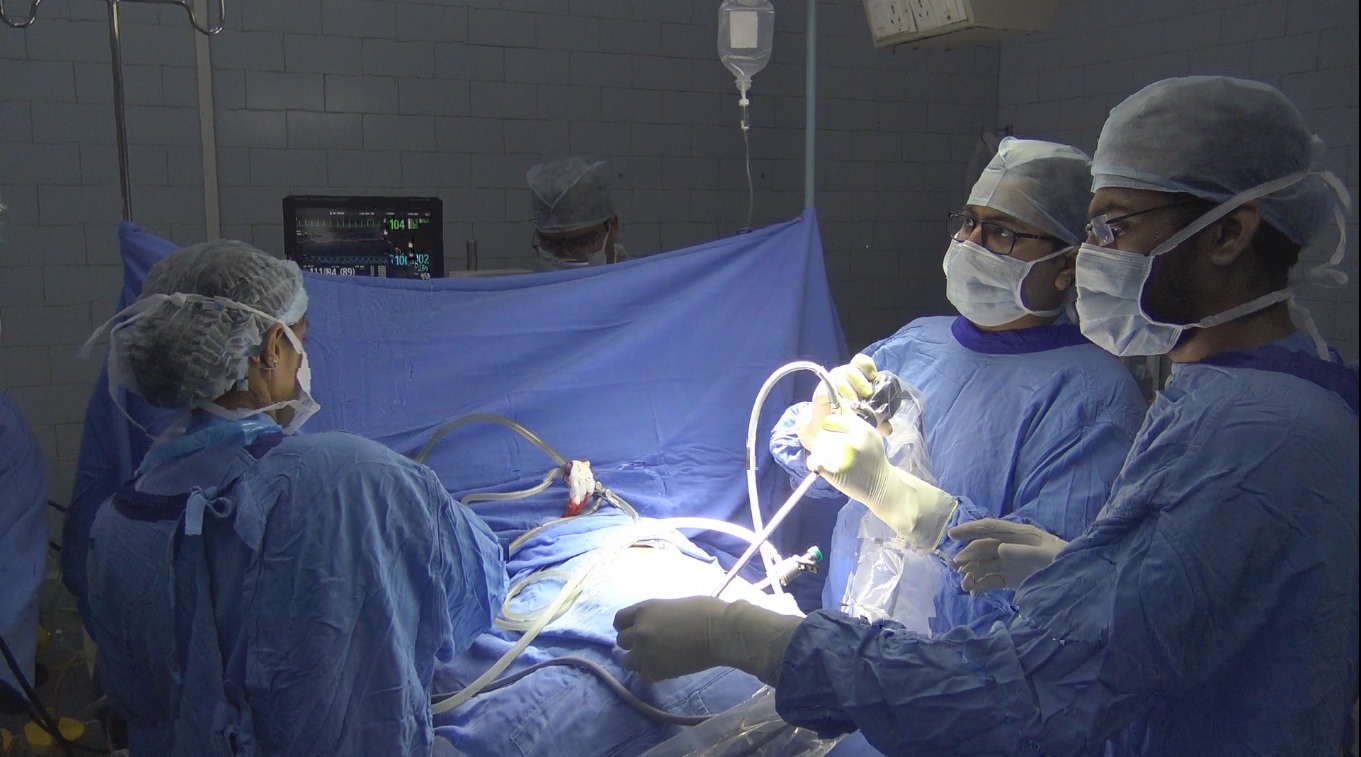

DNB Programme
The National Board of Examinations, New Delhi, an autonomous body directly under the Government of India, award post-graduate and post-doctoral…


TELEMEDICINE
We have been a part of Indian Space Research Organisation (ISRO) telemedicine and tele-education programmes since Sri Sathya Medical Trust..


Papers Published
The doctors and DNB trainees in our hospital have been actively publishing papers in renowned journals and also at various National and Internatio…


FACILITIES
The DNB candidates have access to a very well equipped library all throughout the day. They also have tele-education facilities to share and learn..
Patient Speak
SAI REHABILITATION PROGRAMME
Sai Rehabilitation Programme(SRP) is a unique follow up programme for cardiac patients being implemented by the Sri Sathya Sai Seva Organisations, India, in coordination with the SSSIHMS, Puttaparthi and SSSIHMS,Whitefield. The programme entails following up the post-op cardiac patients in their homes to make sure they are following the regimen prescribed by the hospital regarding their diet and medicines and tests. The programme also helps in creating a support system to the patients in their home towns by organising medical camps, where the patients can get their tests done. The programme also acts as a bridge between the patient and the hospitals. Currently the SRP is being run successfully in 11 states of India namely, Karnataka, Andhra Pradesh, Telangana, Tamil Nadu, Kerala, Odisha, West Bengal, Madhya Pradesh, Maharashtra, Bihar and Jharkhand.



about sssihms
Constructed in six months with a structural area of 320,000 square feet, it is the first super specialty hospital in India to offer world-class tertiary care through state-of-the-art medical technology, totally free of charge to all the patients. This 300 bedded tertiary care hospital is equipped with, 14 operation theatres, one intensive care unit, two cardiac catheterisation laboratories, five in-patient wards and a 24-hour emergency unit. It has a staff of specialists in cardiology, cardiothoracic and vascular surgery, urology, ophthalmology, plastic surgery, orthopaedics, gastroenterology, anaesthesiology, laboratory services and medical imaging. Patients from India and abroad avail the facilities at the hospital.
“I have a task to foster all mankind and ensure for all of them lives full of bliss. I am attached to a work that I love…to remove the suffering of the poor and grant them what they lack ”
Bhagawan Sri Sathya Sai Baba
Contact Us
For treatment related queries please call
080 4710 4600
Treatment Related
080 4710 4600
enquirypg@sssihms.org.in (For treatment and appointment related queries)
General Queries
08555-287388 Extn 1709
publicrelationspg@sssihms.org.in (only for non-treatment/appointment related queries)
Academics Related
academicspg@sssihms.org.in(for queries related to dnb programme)
- INSTRUCTIONS TO PATIENTS
– Please do not visit the hospital without taking a prior appointment with the department you wish to visit
-For taking an appointment you need to send the scanned copies of your medical reports with scanned copy your i-card (Aadhar or Voter’s card) to the following email id. enquirypg@sssihms.org.in
-Please mention clearly in the Subject of your email the name of the department for which you require an appointment. For Ex: Cardiology Appointment Required or Urology Appointment Required etc.-You may mention the choice of the date of your visit for our consideration, but please note that finally your visit date will depend of the availability of slots in our system.
– Please do not send the same e-mail request multiple times or details of the same patient from different e-mail ids. This causes confusion and delays the reply. Moreover such emails may end up in the spam folder.
Bio -Medical Waste Disposal Statistics